DrugAI
Classification of Drug Like molecules using Artificial Neural Network
Sub-Projects
The main aim of the article which I am writing here is to give some brief description about a hobby project of mine called DrugAI. Before reading any further I would recommend you the reader to read and understand about Machine learning and Artificial intelligence. Whereever necessary I will also try to add some quotes and links from internet and lastly I would like to apologize for my english because its not my native language
A few months back I came across Artificial Neural Networks(ANN) and from that day onwards I am thinking of its application in scientific problems. As my educational background is in biology and chemistry I tought of applying ANN in medical related application. The immediate thing which came into my mind was the application of ANN in drug discovery. You may ask Why drug discovery?, because its a multi billion dollar business, it has expensive R&D, takes a longer time for completion(15~30 years) and availability of large amounts of data. The aim of the project is to give scientist or enthusist a tool for the faster classification of drug like molecules (also called as ligand) from a large chemical database using its chemical struture. This process is also called as Ligand based drug design. Ligand is a molecule (eg:glucose) which binds to a target (eg: protein,enzyems etc) resulting in some biological reaction. The idea is that if we can find a chemical molecule whose struture is simliar to that of the naturally seen ligand then it too have similar functions. My best model can classify a model with about 89 percentage validation accuracy . Before explaining my experiment I would mention some techiques and terms. I would recommend you to also refer some other external articles and papers
Artificial Neural Networks
Artificial Neural Networks are relatively crude electronic models based on the neural structure of the brain. The brain basically learns from experience. It is natural proof that some problems that are beyond the scope of current computers are indeed solvable by small energy efficient packages. This brain modeling also promises a less technical way to develop machine solutions. This new approach to computing also provides a more graceful degradation during system overload than its more traditional counterparts.

Simplified Molecular Input Line Entry System(SMILES)
The simplified molecular-input line-entry system or SMILES is a specification in form of a line notation for describing the structure of chemical species using short ASCII strings. SMILES denotes a molecular structure as a graph with optional chiral indications. This is essentially the two-dimensional picture chemists draw to describe a molecule. SMILES describing only the labeled molecular graph (i.e. atoms and bonds, but no chiral or isotopic information) are known as generic SMILES. There are usually a large number of valid generic SMILES which represent a given structure. A canonicalization algorithm exists to generate one special generic SMILES among all valid possibilities; this special one is known as the "unique SMILES". SMILES written with isotopic and chiral specifications are collectively known as "isomeric SMILES". A unique isomeric SMILES is known as an "absolute SMILES". See the following examples[REF 1].
| SMILES | Name | SMILES | Name |
|---|---|---|---|
| CC | ethane | [OH3+] | hydronium ion |
| O=C=O | carbon dioxide | [2H]O[2H] | deuterium oxide |
| C#N | hydrogen cyanide | [235U] | uranium-235 |
| CCN(CC)CC | triethylamine | F/C=C/F | E-difluoroethene |
| CC(=O)O | acetic acid | F/C=C\F | Z-difluoroethene |
| C1CCCCC1 | cyclohexane | N[C@@H](C)C(=O)O | L-alanine |
| c1ccccc1 | benzene | N[C@H](C)C(=O)O | D-alanine |
| Reaction SMILES | Name |
|---|---|
| [I-].[Na+].C=CCBr>>[Na+].[Br-].C=CCI | displacement reaction |
| (C(=O)O).(OCC)>>(C(=O)OCC).(O) | intermolecular esterification |
Bag of words or Bag of N-grams model
Bag of words model is a natural language processing where a document is represented as collection of words and ignoring its sequence. The application of this model includes sentimental analysis, spam-filtering, document classification etc. In the fields of computational linguistics and probability, an n-gram is a contiguous sequence of n items from a given sequence of text or speech.
In this project bag of words model is used to create feature values for SMILES from its individual elements. Consider example for ethane, its SMILE is CC after tokenizing it will become['C', 'C'] and mapped these values to an integer {'C': 0}. For a complex molecule like this CC(=O)NC1=C(N)C=C(C=C1)C([O-])=O its vectorized form will be [0, 4, 4, ..., 1, 0, 0]
DrugAI Project Details and Result
Python is used as the main programming lanugaes. Keras with theano backend is used for making ANN. Scikit-Learn is used for creating n-grams and also for cross validation
Stahl's SMILES dataset were used for this supervised learning. The dataset contains six set of active compounds which includes 128 COX2 Inhibitors,55 Estrogen Receptor Ligands,43 Gelastinase A and General MMP Ligands,17 Neuraminidase Inhibitors,25 p38 MAP Kinase Inhibitors,67 Thrombin Inhibitors. The SMILES data is used as the feature vector and one hot encoding is used for creating target vectors.
Sample Dataset
| SMILES | cox2 | estrogen | gelatinase | neuramidase | kinase | thrombin |
| C[S](=O)(=O)NC1=C(OC2CCCCC2)C=C(C=C1)[N](=O)=O | 1 | 0 | 0 | 0 | 0 | 0 |
| C[S](=O)(=O)NC1=C(OC2=C(F)C=C(F)C=C2)C=C3C(=O)CCC3=C1 | 1 | 0 | 0 | 0 | 0 | 0 |
| C[S](=O)(=O)NC1=C(SC2=C(F)C=C(F)C=C2)C=C3C(=O)CCC3=C1 | 1 | 0 | 0 | 0 | 0 | 0 |
| C[S](=O)(=O)NC1=C(SC2=C(F)C=C(F)C=C2)C=C(C=C1)C(N)=O | 1 | 0 | 0 | 0 | 0 | 0 |
ANN Struture
|
Layers |
Number of Neurons |
Activation Function |
|
1 (Input) |
Extracted Feature Dimension |
Relu |
|
2 |
100 |
Relu |
|
3 |
Dropout Layer ( 20% ) |
|
|
4 |
150 |
Relu |
|
5 (Output) |
6 |
Softmax |
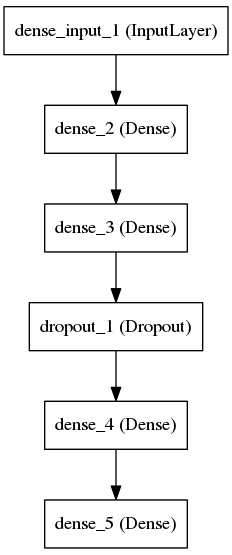
The dataset is first split into training (70%) and testing (30%) set. From the training set the SMILES data were then tokenized into its individual characters , made n-gram of SMILES characters where n=1,2 and3 and then vectorized those n-gram characters. The vectorized values then passed throught the neural network and trained it for certain number of epoch with a batch size of 15. crossvalidation of the testing set was also performed
*All accuracy and loss values in the table are 10-fold cross validated validation accuracy and loss values
Relationship between N-grams and accuracy
|
N-gram Range(Min,Max) Epoch: 20 |
Accuracy |
Loss |
|
(1, 1) |
0.6563 |
0.8954 |
|
(2, 2) |
0.8118 |
0.5373 |
|
(3, 3) |
0.8754 |
0.4455 |
|
(4, 4) |
0.8966 |
0.3852 |
|
(1, 2) |
0.8215 |
0.5054 |
|
(1, 3) |
0.8851 |
0.4050 |
|
(2, 3) |
0.8855 |
0.4169 |
The model showed a direct relationship between the range of n-grams and accuracy. The more number of n-grams the more accurate the model became
Relationship between epoch and accuracy
Two n-grams of range (1,1) and (2,2) were used for this study as expected the number of epochs increased as the accuracy of the model also increased
N-gram (1,1)
|
Epoch n-gram[1, 1] |
Accuracy |
Loss |
|
10 |
0.5549 |
1.1739 |
|
20 |
0.6563 |
0.8954 |
|
40 |
0.7510 |
0.6355 |
|
80 |
0.8065 |
0.4987 |
|
150 |
0.8377 |
0.4725 |
N-gram (2,2)
|
Epoch (2, 2) |
Accuracy |
Loss |
|
10 |
0.7375 |
0.6758 |
|
20 |
0.8118 |
0.5373 |
|
40 |
0.8495 |
0.4975 |
|
80 |
0.8684 |
0.5627 |
|
150 |
0.8770 |
0.6188 |
Possible faults in the model
-
Used less epochs for training because of my inferior computer system (a 8 year old CPU machine)
-
Dosen't optimized the neural network architecure
-
Didn't used higher N-grams
-
Not tried out of sample validation
- Less number of Data
Future Works
-
Use RNN, CNN, SVM etc.
-
Larger dataset
-
Use other chemical data formats such as International Chemical Identifier
-
Predict strutures instead of classification